
.svg.png)





The U.S. Food and Drug Administration (FDA) recently made what some believe is a controversial move to ensure laboratory-developed tests’ (LDTs) safety and effectiveness. In a news release dated April 29, 2024, the FDA announced its final rule amending its regulations to make explicit that LDTs are IVDs and are classified as devices under the Federal Food, Drug, and Cosmetic Act (FD&C Act). The FDA further elaborated that laboratories are not immune from this rule as manufacturers of in vitro diagnostic products (IVDs).
Under this new rule, as defined by the FDA, LDTs are IVDs “intended for clinical use and designed, manufactured and used within a single clinical laboratory that meets certain regulatory requirements.” IVDs are frequently used to collect human specimens from inside the body to diagnose certain conditions. IVDs, including LDTs, can measure or detect various substances in human blood, saliva, or tissue to determine what diseases to treat or assist practitioners in devising a plan for ongoing patient care.
LDTs are often developed when no other test is available or appropriate to meet the needs of a specific patient population. Whether manufactured commercially or developed in the lab, clinical laboratory tests steer medical decisions most of the time (more than 70%). This makes them a “critical component” of medicine and patient care, with LDTs being considered the “gold standard” in diagnosing and monitoring many conditions. However, until now, only IVDs had to undergo an FDA regulatory submission process. This new rule expands regulation to cover LDTs.
Some believe this broadened oversight is long overdue. The Center for Science in the Public Interest (CSPI) published an article in February 2024 citing the dangers of under-regulation, specifically concerning LDTs. The organization recognizes the overall relevance and importance of LDTs but also points out, “Due to lax oversight, some LDTs are unreliable or make claims based on weak evidence, resulting in wasted time and resources and even significant harm to patients.”
CSPI reported on a lawsuit it filed against EpicGenetics in 2023 for false claims regarding the accuracy of its fibromyalgia tests and a so-called “Immune Deficiency Disease.” Additionally, CSPI noted that even with oversight per the Clinical Laboratory Improvement Amendments (CLIA), administered by an agency other than the FDA, the FDA released 20 case studies revealing faulty LDTs in 2015 as evidence of why its oversight is necessary. As such, this isn’t the first time the FDA has tried to step in to regulate LDTs. In 2014, a draft guidance released by the FDA required premarket reviews before high-risk LDTs could be marketed. This guidance was later withdrawn in 2017 due to pushback from various trade associations, academic medical centers, and Congress.
Once again, not everyone is happy with the FDA’s newest guidance, and they have taken their objections to court. Organizations, including the American Clinical Laboratory Association, oppose the rule, arguing that it “exceeds FDA’s lawful authority and is arbitrary and capricious and contrary to law.” As a result, they are suing the federal entity, requesting that the rule be set aside and vacated and that the FDA, the U.S. Department of Health and Human Services (HHS), and other governing individuals and authorities be barred from implementing and enforcing the rule.
The American Hospital Association (AHA) previously voiced its opinion to Congress, alleging overreach by the FDA. While the organization supports the need for additional oversight of developing and using “some” LDTs and IVDs offered as LDTs, it is against the FDA’s device regulations applying to hospital and health system LDTs. The AHA calls the move “misguided,” stating that the tests are not devices but diagnostic tools that are a part of “essential patient care.” The opposition continued, voicing concerns about the accessibility of these “critical tests” under the FDA’s new rule. Additionally, the AHA argued that the latest device regulatory framework would “undermine” or “stifle” innovation in hospital and health system laboratory medicine.
In light of the backlash surrounding the FDA’s final rule, we had Managing Practice Director Gary Bergman weigh in with his thoughts regarding the new regulation. Bergman’s vote was in favor of the new oversight, stating, “I support LDTs being regulated as devices to ensure that they meet the same safety, efficacy, and quality standards as commercially available diagnostic tests, thereby protecting public health, providing patient confidence, and maintaining the integrity of clinical decision-making.”
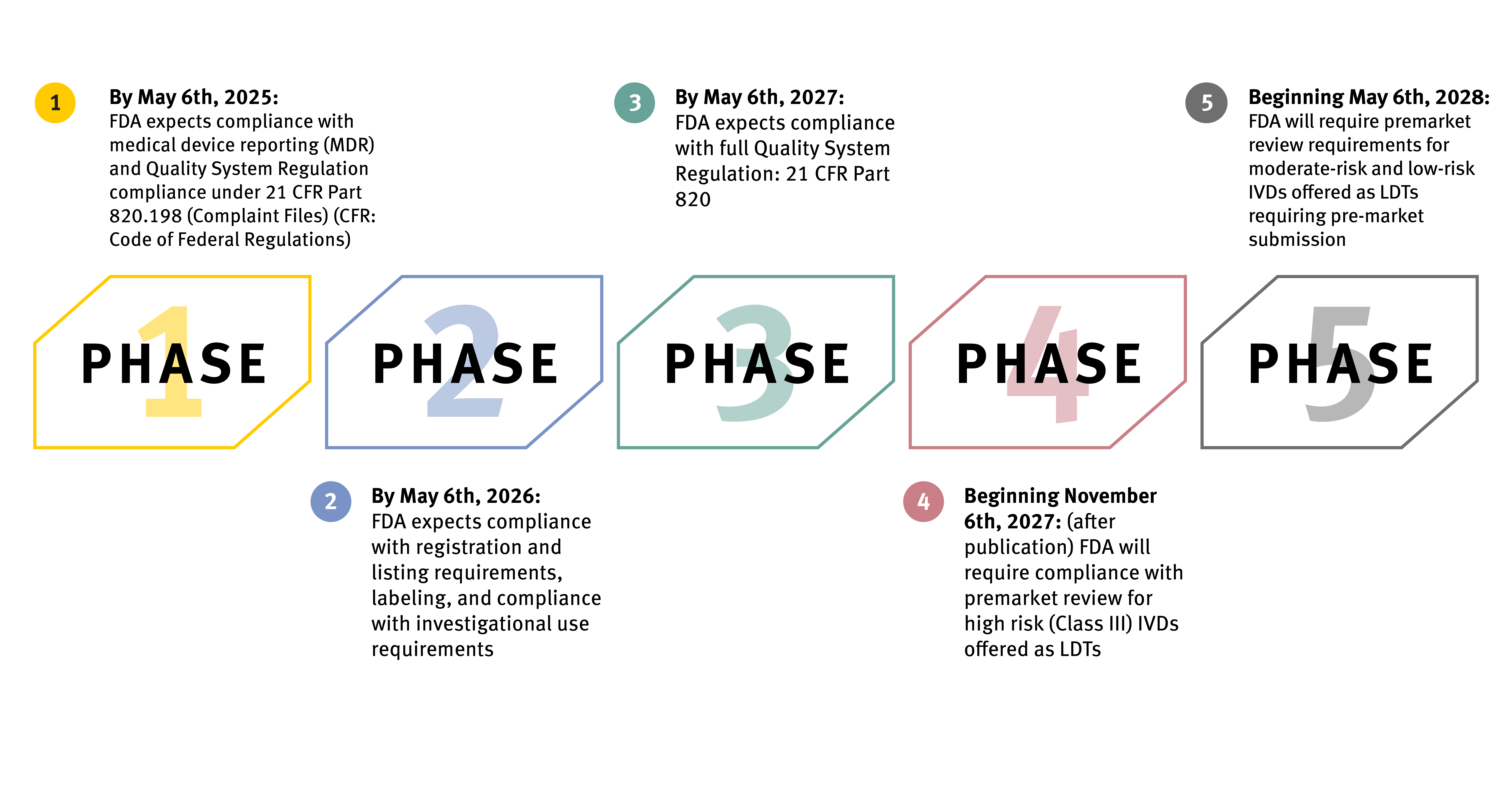
If allowed to proceed, the final rule affords a four-year phaseout approach that will take place in five stages. The FDA expects LDTs to fall under the specific regulatory requirements outlined at each stage and meet compliance to avoid enforcement and applicable penalties. Specific tests fall outside the scope of the policy, including donor screening tests, tests intended for emergencies, direct-to-consumer tests, and tests for the exclusive use of public health surveillance.
The FDA intends for enforcement to be fully implemented by May 6, 2028, with the first stage beginning one year after the final rule’s publication date.
Oxford can help keep your organization updated on recent and future events and ensure compliance with current and changing regulations. We have the knowledge and skills to ensure a smooth transition over the next four years so that you can continue to provide the best support to health systems, medical practitioners, and patients.
